 MENU
MENU 

Gokul Ravi earns Innovation Award in ICCAD Quantum Computing for Drug Discovery Challenge

Prof. Gokul Ravi and his coauthors have received the Innovation Award in the first-ever Quantum Computing for Drug Discovery Challenge at the 2023 IEEE/ACM International Conference on Computer-Aided Design (ICCAD). The team was recognized for their exceptional work on the extension of classical computing techniques to variational quantum algorithms, as described in their paper “CAFQA: A Classical Simulation Bootstrap for Variational Quantum Algorithms.”
ICCAD, taking place October 29-November 2 in San Francisco, CA, is the leading global conference for computer-aided design. It aims to promote the advancement of the field and related technology by providing a forum for professionals to share new ideas and research topics.
This year was the first iteration of the ICCAD Quantum Computing for Drug Discovery Challenge, a multi-month competition that tasks researchers with addressing drug discovery-related problems that require quantum algorithms. The challenge is open to teams across the world; this year, 73 teams from 12 different countries competed, and just one was selected to receive the Innovation Award.
The winning solution developed by Ravi and his coauthors is CAFQA, a Clifford Ansatz For Quantum Accuracy, a classical computing method that bootstraps variational quantum algorithms in order to more accurately estimate the ground state energy of molecules. Quantum computing is essential in drug discovery and molecular chemistry more broadly, as it allows researchers to study and understand molecules at the most fundamental level, where quantum mechanical properties can be observed.
“The discovery of novel drugs is heavily dependent on a better understanding of chemical reactions and how different molecules interact with each other in different environments,” said Ravi. “Quantum computing is critical in this area because of its utility in understanding the behavior of these molecules at the subatomic level.”
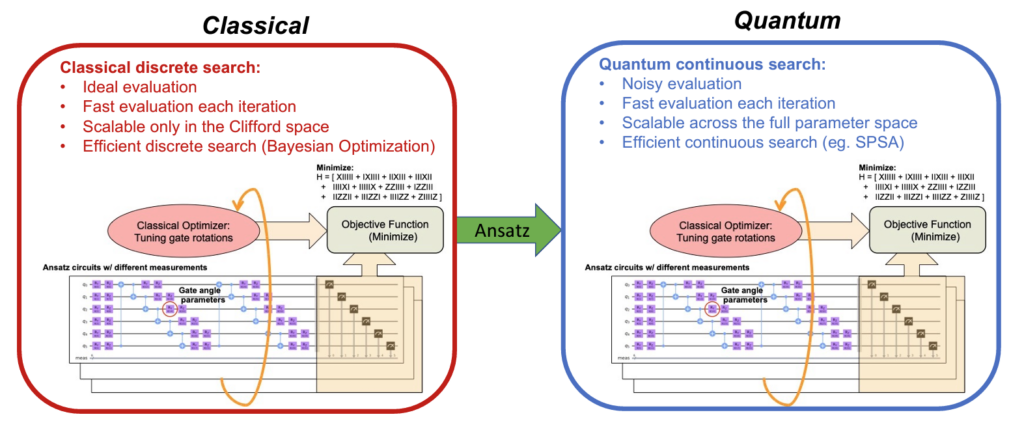
CAFQA harnesses the simplicity and efficiency of classical computing methods to run variational quantum algorithms. This enables significantly faster, more accurate estimations of ground state energy, which is required in order to break up molecules into their subatomic components. In addition to its improvements in speed and accuracy, its reliance on classical simulation methods means that CAFQA is much easier to run on quanum devices, which are hampered by qubit noise and high costs.
In sum, CAFQA is a powerful tool that allows researchers across the quantum computing and molecular chemistry disciplines to better break down and understand the fundamental functions, behaviors, and interactions of molecules, an essential part of developing new drugs.
“CAFQA is a vital step forward in the important future domain of hybrid quantum-classical computing,” said Ravi.